Мышечные дистрофии. Мышечная дистрофия у детей: симптомы, лечение, диагностика, причины
Мышечная дистрофия Дюшенна – это генетическое заболевание, связанное с нарушением строения мышечных волокон. Мышечные волокна при этой болезни, в конце концов, распадаются, и теряется способность к передвижению. Мышечная дистрофия Дюшенна передается сцеплено с полом, болеют лица мужского пола. Проявляет себя уже в детском возрасте. Помимо мышечных нарушений, заболевание приводит к скелетным деформациям, может сопровождаться дыхательной и сердечной недостаточностью, умственными и эндокринными нарушениями. Радикального способа лечения, позволяющего искоренить болезнь, пока нет. Все существующие меры являются лишь симптоматическими. Довольно редко больным удается пережить рубеж 30 лет. Эта статья посвящена причинам, симптомам, диагностике и лечению мышечной дистрофии Дюшенна.
Заболевание впервые описано в 1861 году (по другим данным – 1868 году) французским невропатологом и носит его имя. Встречается не так уж и редко: 1 случай на 3500 новорожденных детей. Из всех известных медицине мышечных дистрофий является наиболее распространенной.

В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Заболевание наследуется по рецессивному типу, сцепленному с Х хромосомой. Что это означает? Поскольку все гены человека парные, то есть дублируют друг друга, то для того, чтобы появились патологические изменения в организме при наследственном заболевании, необходимо, чтобы генетический дефект возник в одной хромосоме или аналогичных участках обеих хромосом. Если заболевание возникает только при мутациях в обеих хромосомах, то такой тип наследования называют рецессивным. Когда же генетическая аномалия выявляется только в одной хромосоме, но болезнь все равно развивается, такой тип наследования называют доминантным. Рецессивный тип возможен только при одновременном поражении идентичных хромосом. Если вторая хромосома будет «здорова», то заболевание не возникнет. Именно поэтому мышечная дистрофия Дюшенна является уделом лиц мужского пола, потому что они имеют в генетическом наборе одну Х хромосому, а вторую (парную) – У. Если мальчику попадается «поломанная» Х хромосома, то у него обязательно возникает болезнь, потому что здоровой хромосомы у него просто нет. Для того, чтобы мышечная дистрофия Дюшенна возникла у девочки, необходимо совпадение в ее генотипе двух патологических Х хромосом, что практически маловероятно (в таком случае папа девочки должен быть болен, а у мамы в генетическом наборе должна содержаться дефектная Х хромосома). Девочки выступают лишь носителями заболевания и передают его своим сыновьям. Конечно, часть случаев заболевания не является результатом передачи по наследству, а возникает спорадически. Это означает появление мутации в генетическом наборе ребенка спонтанно. Вновь появившаяся мутация может быть передана по наследству (при условии сохранения способности к размножению).
Симптомы заболевания
Мышечная дистрофия Дюшенна всегда заявляет о себе до 5-летнего возраста. Наиболее часто первые симптомы возникают еще до 3-х лет. Все патологические проявления заболевания можно разделить на несколько групп (в зависимости от характера изменений):
- поражение скелетной мускулатуры;
- деформации скелета;
- поражение сердечной мышцы;
- умственные нарушения;
- эндокринные расстройства.
Поражение скелетных мышц

Поражение мышечной ткани является основным проявлением заболевания. Оно становится причиной генерализованной мышечной слабости. Начальные симптомы подкрадываются незаметно.
Рождаются дети без особых отклонений. Однако их двигательное развитие отстает в темпах по сравнению со сверстниками. Такие дети менее активные и подвижные в двигательном плане. Пока ребенок совсем мал, это часто связывают с особенностями темперамента и не обращают внимания на начальные изменения.
Явные признаки возникают с началом ходьбы. Дети часто падают и передвигаются на пальцах (на носочках). Следует отметить, что эти нарушения интерпретируют не при первых шагах ребенка, потому что прямохождение вначале для всех детей сопряжено с падениями и неуклюжестью. Когда большинство ровесников уже вполне уверенно передвигается, мальчики с мышечной дистрофией Дюшенна упорно продолжают падать.
Когда ребенок научится разговаривать, он начинает жаловаться на слабость и быструю утомляемость, непереносимость физических нагрузок. Бег, лазанье, прыжки и другие любимые виды активной деятельности детей для ребенка с мышечной дистрофией Дюшенна не привлекательны.
Походка таких детей напоминает утиную: они как бы переваливаются с ноги на ногу.
Своеобразным проявлением заболевания является симптом Говерса. Он заключается в следующем: при попытке ребенка подняться с колен, корточек, с пола он пользуется руками, чтобы помочь слабым мышцам ног. Для этого он опирается руками на себя, «взбираясь по лесенке, по самому себе».
Мышечная дистрофия Дюшенна имеет восходящий тип мышечной слабости. Это означает, что сначала слабость проявляет себя в ногах, затем распространяется на таз и туловище, потом на плечи, шею и, в конце концов, на руки, дыхательную мускулатуру и голову.
Несмотря на то, что при этом заболевании мышечные волокна подвергаются разрушению и развитию атрофии, внешне некоторые мышцы могут выглядеть вполне нормальными или даже накаченными. Развивается так называемая псевдогипертрофия мышц. Чаще всего этот процесс заметен в икроножных, ягодичных и дельтовидных мышцах, мышцах языка. Место распавшихся мышечных волокон занимает жировая ткань, именно поэтому создается эффект хорошей развитости мускулатуры, что на проверку оказывается совсем не так.
Атрофический процесс в мышцах всегда симметричный. Восходящее направление процесса приводит к возникновению «осиной» талии, «крыловидных» лопаток (лопатки отстают от туловища, словно крылья), симптома «свободных надплечий» (когда голова как бы проваливается в плечи при попытке поднять ребенка под мышки). Лицо гипомимично, губы могут утолщаться (замена мышц жировой и соединительной тканью). Псевдогипертрофия языка становится причиной речевых расстройств.
Разрушение мышц сопровождается развитием мышечных контрактур и укорочением сухожилий (хорошо заметно на примере ахиллового сухожилия).
Сухожильные рефлексы (коленный, ахиллов, с бицепса, с трицепса и так далее) постепенно снижаются. Мышцы на ощупь плотные, но безболезненные. Мышечный тонус обычно снижается.
Постепенное прогрессирование мышечной слабости приводит к тому, что к 10-12 годам многие дети утрачивают способность самостоятельно передвигаться и нуждаются в инвалидном кресле. Способность стоять сохраняется, в среднем, до 16 лет.
Отдельно следует сказать о вовлечении в патологический процесс дыхательной мускулатуры. Это наблюдается после подросткового периода. Слабость диафрагмы и других мышц, участвующих в акте дыхания, приводит к постепенному снижению жизненной емкости легких и объемов вентиляции. По ночам это особенно заметно (появляются приступы удушья), поэтому у детей могут возникать страхи перед сном. Формируется дыхательная недостаточность, которая усугубляет течение интеркуррентных инфекций.
Деформации скелета

Это сопутствующие мышечным изменениям симптомы. У детей постепенно формируются усиление поясничного изгиба (лордоза), искривление грудного отдела позвоночника в сторону (сколиоз) и сутулость (кифоз), меняется форма стопы. Со временем развивается диффузный остеопороз. Эти симптомы еще больше способствуют ухудшению двигательных нарушений.
Поражение сердечной мышцы
Является обязательным симптомом мышечной дистрофии Дюшенна. У больных развивается кардиомиопатия (гипертрофическая или дилатационная). Клинически это проявляет себя нарушениями сердечного ритма, перепадами артериального давления. Границы сердца увеличиваются, однако такое большое сердце имеет малые функциональные возможности. В конце концов, формируется сердечная недостаточность. Сочетание выраженной сердечной недостаточности с дыхательными расстройствами на фоне присоединившейся инфекции может быть причиной смертельного исхода у больных с мышечной дистрофией Дюшенна.
Умственные нарушения
Это не обязательный, но возможный признак заболевания. Он связан с дефицитом особой формы дистрофина – аподистрофином, содержащимся в головном мозге. Нарушения интеллекта варьируются от незначительных до степени идиотии. При этом выраженность умственных нарушений никоим образом не связана со степенью мышечных расстройств. Социальная дезадаптация из-за невозможности свободно передвигаться и посещать детские учреждения (садики, школы) способствует усугублению когнитивных расстройств.
Эндокринные расстройства
Встречаются у 30-50% больных. Могут быть довольно разнообразными, но чаще всего это ожирение с преимущественным отложением жира в области молочных желез, бедер, ягодиц, плечевого пояса, недоразвитие (или нарушение функции) половых органов. Больные часто имеют низкий рост.
Мышечная дистрофия Дюшенна неуклонно прогрессирует. К 15-20 годам почти все больные не в состоянии себя обслуживать самостоятельно ввиду обездвиженности. В конце концов, присоединяются бактериальные инфекции (органов дыхания и мочевыделительной сферы, инфицированные пролежни при недостаточном уходе), которые на фоне сердечной и дыхательной недостаточности приводят к смертельному исходу. Мало кто из больных переживает рубеж в 30 лет.
Диагностика

Диагностика мышечной дистрофии Дюшенна основывается на нескольких видах исследований, основным из которых является генетический тест (ДНК-диагностика).
Только обнаружение дефекта Х хромосомы в том участке, который отвечает за синтез дистрофина, достоверно подтверждает диагноз. До проведения такого анализа диагноз является предварительным.
Из других методов исследования могут применяться:
- определение активности креатинфосфокиназы (КФК). Этот фермент отражает гибель мышечных волокон. Его концентрация при мышечной дистрофии Дюшенна превышает норму в десятки и сотни раз до 5-летнего возраста. Позже уровень фермента постепенно снижается, потому что часть мышечных волокон уже необратимо разрушена;
- электромиография. Этот метод позволяет подтвердить тот факт, что в основе заболевания лежат первичные изменения мышц, а нервные проводники при этом совершенно интактны;
- биопсия мышц. С ее помощью определяют содержание белка дистрофина в мышце. Однако в связи с усовершенствованием генетической диагностики в последние десятилетия эта травматичная процедура отошла на второй план;
- дыхательные пробы (исследование жизненной емкости легких), ЭКГ, УЗИ сердца. Эти методы не используются для установления диагноза, но необходимы для выявления патологических изменений со стороны дыхательной и сердечно-сосудистой систем для того, чтобы скорригировать имеющиеся нарушения.
Выявление больного ребенка в семье означает, что в генотипе матери имеется патологическая Х хромосома. В редких случаях мать может быть здоровой, если мутация возникла у ребенка случайно. Наличие дефектной Х хромосомы несет в себе риск для последующих беременностей. Поэтому такие семьи должен консультировать генетик. При наступлении повторных беременностей родителям предлагают пренатальную диагностику, то есть исследование генотипа еще не родившегося ребенка с целью исключения наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Для исследования понадобятся клетки плода, которые получают с помощью различных процедур на разных сроках беременности (например, биопсия хориона, амниоцентез и другие). И хотя эти медицинские манипуляции несут в себе определенный риск для беременности, они позволяют точно ответить на вопрос: имеется ли у плода генетическое заболевание.
Лечение
Мышечная дистрофия Дюшенна является в настоящее время неизлечимым заболеванием. Можно помочь ребенку (взрослому) продлить время двигательной активности, с помощью различных способов поддерживая мышечную силу, компенсируя изменения со стороны сердечно-сосудистой и дыхательной систем.
Несмотря на это, прогнозы ученых в отношении полного излечения от этого заболевания довольно оптимистичны, поскольку уже сделаны первые шаги в этом направлении.
В настоящее время, для лечения мышечной дистрофии Дюшенна из медикаментозных препаратов используют:
- стероиды (при регулярном применении они позволяют уменьшить мышечную слабость);
- β-2-адреномиметики (также временно придают силу мышцам, но не замедляют прогрессирование заболевания).
Применение β-2-адреномиметиков (Альбутерол, Формотерол) не имеет статистически достоверного признания, поскольку существует небольшой опыт их использования при данной патологии. Контроль изменений состояния здоровья у группы пациентов, применявших эти препараты, проводился в течение одного года. Поэтому утверждать, что они работают более длительное время, нет возможности.
Основой лечения сегодня являются стероиды. Считается, что их использование позволяет какое-то время сохранять мышечную силу, то есть они могут затормозить прогрессирование болезни. Кроме того, доказано, что стероиды уменьшают риск возникновения сколиоза при мышечной дистрофии Дюшенна. Но все же возможности этих препаратов ограничены, и заболевание будет неуклонно прогрессировать.
 Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Из медикаментозных препаратов также при мышечной дистрофии Дюшенна применяют сердечные средства (антиаритмические, метаболические, ингибиторы ангиотензинпревращающего фермента). Они позволяют бороться с кардиологическими аспектами заболевания.
Из немедикаментозных методов лечения значимую роль играют физиотерапия и ортопедическая помощь. Физиотерапевтические методики позволяют дольше, чем без их применения, сохранять гибкость и подвижность суставов, поддерживают мышечную силу. Доказано, что умеренная физическая активность благоприятно влияет на течение заболевания, а вот бездействие и постельный режим наоборот способствуют еще более быстрому прогрессированию заболевания. Поэтому необходимо как можно дольше поддерживать посильную физическую деятельность, даже после того, как больной «пересел» в инвалидное кресло. Показаны регулярные курсы массажа. На самочувствие больного положительно влияет плавание.
Ортопедические приспособления позволяют значительно облегчить жизнь больного. Их перечень довольно широк и разнообразен: это и различного рода вертикализаторы (помогают сохранять положение стоя), и приспособления для самостоятельного вставания, и инвалидные кресла-коляски с электрическим приводом, и специальные шины для устранения контрактур в голени (используются даже ночью), и корсеты для позвоночника, и длинные шины для ног (колено-голеностопные ортезы), и многое другое.
Когда заболевание поражает и дыхательные мышцы, и самостоятельное дыхание становится неэффективным, то возможно применение аппаратов искусственной вентиляции легких различной модификации.
И все же даже использование всех этих мер в комплексе не позволяет побороть заболевание. На сегодняшний день, есть ряд перспективных направлений исследований, которые, возможно, станут прорывом в лечении мышечной дистрофии Дюшенна. К наиболее распространенным среди них относят:
- генную терапию (введение «правильного» гена с помощью вирусных частиц, доставку генетических конструкций в составе липосом, олигопептидов, полимерных носителей и другие);
- регенерацию мышечных волокон с помощью стволовых клеток;
- трансплантацию миогенных клеток, которые способны к синтезу нормального дистрофина;
- пропуск экзонов (с помощью антисмысловых олигорибонуклеотидов) в качестве попытки замедлить прогрессирование заболевания и смягчить его течение;
- замену дистрофина на другой белок атрофин, ген которого расшифрован. Методика опробована на мышах и дала положительные результаты.
Каждая из новых разработок несет в себе надежду для больных с мышечной дистрофией Дюшенна на полное выздоровление.
Таким образом, мышечная дистрофия Дюшенна – это генетическая проблема лиц мужского пола. Болезнь характеризуется прогрессирующей мышечной слабостью из-за разрушения мышечных волокон. В настоящее время является неизлечимым заболеванием, однако многие ученые мира трудятся над созданием радикального способа борьбы с ним.
Мультипликационный фильм «Мышечная дистрофия Дюшенна», англ. озвучка, субтитры на русском языке:
Мышечной дистрофией фактически называют группу наследственных заболеваний, характеризуемых прогрессирующей симметричной атрофией скелетных мышц, протекающей без болей и потери чувствительности в конечностях. Парадоксально, но пораженные мышцы могут увеличиваться в размерах из-за роста соединительной ткани и жировых отложений, создавая ложное впечатление крепких мышц.
До сих пор не существует средства, излечивающего мышечную дистрофию. Различают четыре основных вида этой патологии. Наиболее часто встречается мышечная дистрофия Дюшенна (50% всех случаев). Обычно заболевание начинается в раннем детстве и приводит к смерти к 20 годам. Мышечная дистрофия Беккера развивается медленнее, больные живут более 40 лет. Плече-лопаточно-лицевая и конечностно-поясная дистрофии, обычно не влияют на продолжительность жизни.
Причины возникновения
Развитие мышечной дистрофии. обусловлено различными генами. Мышечная дистрофия Дющенна и мышечная дистрофия Беккера вызываются генами, находящимися в половой хромосоме, и заболевают ими только мужчины. Плече-лопаточно-лицевая и конечностно-поясничная дистрофии не связаны с половыми хромосомами; ими заболевают и мужчины, и женщины.
Симптомы
Все разновидности мышечной дистрофии вызывают прогрессирующую атрофию мышц.
Диагностика
Врач осматривает ребенка, задает вопросы о заболеваниях у членов семьи и назначает определенные исследования. Если кто-либо из родственников болел мышечной дистрофией, врач выясняет, как протекала у него дистрофия. Анализируя полученные данные, можно прогнозировать, что ожидает ребенка. Если в семье не было больных мышечной дистрофией, электромиография позволит оценить функционирование нервов в пораженных мышцах и установить наличие мышечной дистрофии; исследование кусочка мышечной ткани () может показать клеточные изменения и наличие жировых отложений.
В медицинских центрах, оснащенных самым современным оборудованием для проведения молекулярно-биологических и иммунологических исследований, могут с точностью определить, будет ли ребенок страдать мышечной дистрофией. В этих центрах могут также провести обследование родителей и родственников на наличие у них генов, определяющих развитие мышечной дистрофии Дюшенна и мышечной дистрофии Беккера.
Виды заболевания
По степени тяжести заболевания и времени его появления различают:
Дистония Дюшенна проявляет себя в раннем возрасте (между 3 и 5 годами). Больные дети ходят вразвалку, с трудом поднимаются по лестнице, часто падают, не могут бегать. Когда они поднимают руки, лопатки у них «отстают» от туловища - этот симптом получил название «крыловидных лопаток». Обычно ребенок с мышечной дистрофией к 9-12 годам оказывается прикованным к инвалидному креслу. Прогрессирующая слабость сердечной мышцы приводит к смерти от внезапно наступившей сердечной недостаточности, дыхательной недостаточности или инфекции.
Хотя дистрофия Беккера имеет много общего с дистрофией Дюшенна, развивается она гораздо медленнее. Симптомы появляются в возрасте около 5 лет, но и после 15 лет больные дети обычно еще сохраняют способность ходить, а иногда и значительно позже.
Плече-лопаточно-лицевая дистрофия развивается медленно, течение ее относительно доброкачественное. Чаще заболевание начинается до 10 лет, но может появиться в раннем подростковом возрасте. Дети, у которых впоследствии обнаруживается эта патология, в младенческом возрасте плохо сосут; когда они становятся старше, им не удается складывать губы как для свистка, поднимать руки выше головы. У больных детей лица отличаются малоподвижностью при смехе или плаче, иногда отмечают мимику, отличную от нормальной.
Действия пациента
Если вы беспокоитесь, что у вашего ребенка, возможно, развивается мышечная дистрофия, вы можете принести фотографии или видеозаписи, которые иллюстрируют ваши конкретные проблемы. Возьмите с собой родственника или друга, который также выслушает информацию, предоставленную врачом.
Лечение
До сих пор нет средства, которое бы могло остановить прогрессирование атрофии мышц при мышечной дистрофии. Однако ортопедические устройства, а также лечебная физкультура, физиотерапия и хирургия для коррекции контрактур могут на некоторое время сохранить подвижность ребенка или подростка.
Членам семей, в которых были случаи заболевания мышечной дистрофией, следует обратиться за медико-генетической консультацией с тем, чтобы узнать, существует ли риск передачи заболевания будущему ребенку.
Осложнения
Некоторые виды мышечной дистрофии сокращают продолжительность жизни человека, часто влияют на мышцы, связанные с дыханием. Даже с помощью улучшенного механического дыхания люди, у которых мышечная дистрофия Дюшенна - наиболее распространенный тип мышечной дистрофии - как правило, умирают от дыхательной недостаточности, не дожив до 40 лет.
Многие виды мышечной дистрофии могут также снизить эффективность сердечной мышцы. Если болезнью затронуты мышцы, связанные с глотанием могут возникнуть проблемы с питанием.
Так как мышечная слабость прогрессирует, подвижность становится проблемой. Многим людям, у которых развивается мышечная дистрофия, возможно, потребуется использовать инвалидное кресло. Тем не менее, длительная неподвижность суставов, связанная с использованием инвалидной коляски, может ухудшить контрактуру, при которой конечности разворачиваются во внутреннем положении и фиксируются в нем.
Контрактуры могут также играть роль в развитии сколиоза, став причиной искривления позвоночника, что еще больше снижает эффективность работы легких у людей, которые больны мышечной дистрофией.
Профилактика
Поскольку респираторные инфекции могут стать проблемой в более поздних стадиях мышечной дистрофии, важно сделать прививку от пневмонии, а также регулярно получать прививки от гриппа.
![]()
Описание:
Мышечная дистрофия представляет собой группу хронических наследственных заболеваний скелетных мышц человека, проявляющихся слабостью и дегенерацией мышц. Существует девять различных форм мышечной дистрофии. Они различаются по таким характеристикам, как возраст, в котором начинается заболевание, локализация пораженных мышц, выраженность мышечной слабости, скорость прогрессирования дистрофии и тип ее наследования. Чаще всего встречаются две формы: мышечная дистрофия Дюшенна и миотоническая мышечная дистрофия.
Симптомы:
Дюшенна дистрофия. Х-хромосомная рецессивная мутация дистрофин-гена. Клинические признаки: начало в возрасте до 5 лет; прогрессирующая слабость мышц тазового и плечевого пояса; неспособность ходить после 12 лет; кифосколиоз; дыхательная недостаточность в возрасте 20-30 лет. Вовлечение других систем органов: ; снижение интеллекта.
Беккера дистрофия. Х-хромосомная рецессивная мутация дистрофин-гена. Клинические признаки: начало в раннем или позднем возрасте; медленно прогрессирующая слабость мышц тазового и плечевого пояса; сохранение способности ходить после 15 лет; дыхательная недостаточность после 40 лет. Вовлечение других систем органов: кардиомиопатия.
Миотоническая дистрофия. Аутосомно- доминантный; расширение нестабильного участка ДНК хромосомы 19ql3,3. Клинические признаки: начало в любом возрасте; медленно прогрессирующая слабость мышц век, лица, шеи, дистальных мышц конечностей; миотония. Вовлечение других систем органов: нарушение сердечной проводимости; психические нарушения; , лобная ; гонад.
Плече-лопаточно-лицевая дистрофия.
Аутосомно-доминантный; часто мутации хромосомы 4q35. Клинические признаки: начало в возрасте до 20 лет; медленно прогрессирующая мышечная слабость лицевой области, плечевого пояса, тыльного сгибания стопы. Вовлечение других систем органов: гипертензия; глухота.
Плечевого и тазового пояса (возможны несколько заболеваний). Аутосомно-рецессивный или доминантный. Клинические признаки: начало с раннего детства до среднего возраста; медленно прогрессирующая слабость мышц плечевого и тазового пояса. Вовлечение других систем органов: кардиомиопатия.
Глазо-глоточная дистрофия. Аутосомно-доминантный (Французская Канада или Испания). Клинические признаки: начало в 50-60 лет; медленно прогрессирующая слабость мышц: наружных глазных, век, лица и глотки; крикофарингеальная ахалазия. Вовлечение других систем органов: церебральные, глазные.
Врожденная дистрофия. Включает несколько заболеваний, в том числе типы Фукуяма и церебро-окулярная дисплазия). Аутосомно-рецессивный. Клинические признаки: начало при рождении; гипотония, задержка развития; в одних случаях - ранняя дыхательная недостаточность, в других - более благоприятное течение болезни.
Причины возникновения:
Заболевание вызывается аутосомно-доминантным геном с резко варьирующей экспрессивностью (возможность риска передачи на родственников 1-й степени составляет 50%). Заболевание вызывается амплификацией, т. е. повышением числа CTG - триплетов в определенном локусе 19 хромосомы (тип 1 миотонической дистрофии) или CCTG в 3 хромосоме (тип2 миотонической дистрофии). Тип 2 миотонической дистрофии изучен слабо. Считается, что он встречается только в 2% случаев (но может быть и значительно чаще); не взаимосвязан с типом 1; скорее всего не является причиной врожденных форм дистрофии, когда носителем является мать. Для 1 типа доказано, что число повторов нуклеотидов увеличивается при передаче мутации из поколения в поколение. Тяжесть заболевания четко коррелирует с численностью этих повторов. Наибольшее их число определяется при врожденной тяжелой форме заболевания . Выявленный механизм объясняет феномен антиципации - утяжеления и все более раннего начала болезни в нисходящих поколениях. К примеру, если генетический анализ показал, что родитель имеет определенное число повторов CTG, то у его ребенка обнаружится еще большее количество повторов этого триплета.
Лечение:
На сегодняшний день не существует способов предотвратить или замедлить прогрессирование данного заболевания. Терапия направлена главным образом на борьбу с осложнениями, такими, как деформация позвоночника, развивающаяся вследствие слабости мышц спины, или предрасположенность к пневмониям, обусловленная слабостью дыхательных мышц. Фенитоин, прокаинамид, хинин применяются в лечении миотонии, но требуется осторожность у больных с заболеваниями сердца (опасность ухудшения сердечной проводимости). Имплантация водителя сердечного ритма необходима больным с синкопе или сердечной блокадой. При лечении сердечных нарушений рекомендован препарат фенигидин. Применение ортопедических аппаратов может укрепить «висячие» стопы, стабилизировать голеностопные суставы, уменьшить частоту падений. Хорошо подобранная тренировка также может оказать положительное влияние на течение данного заболевания. При наличии атрофии применяют анаболические стероиды (ретаболил, неробол), общеукрепляющую терапию. В тех случаях, когда имеется значительно выраженная миотоническая симптоматика, назначают курсы дифенина по 0,03-0,05 г 3 раза в день, длительностью 2-3 нед. Предполагают, что дифенин оказывает угнетающее действие на синаптическую проводимость и снижает посттетаническую активность в мышцах. При повышенной сонливости, нередко сопровождающей миотоническую дистрофию, положительный эффект наблюдается при приеме селегилина. Рекомендован также прием некоторых биологически активных добавок: кофермента Q10 (100 мг/ день), витамина Е (200 МЕ/день) и селена (200 мкг/день), лецитина (20 г/день).
Эффективное излечение от данного заболевания возможно только при помощи генной терапии, которая сейчас интенсивно развивается. Многочисленные эксперименты показывают улучшение состояния мышечных волокон при лечении некоторых форм мышечной дистрофии. При дистрофиях Дюшена и Беккера наблюдается недостаточная выработка мышечного белка дистрофина. Ген, ответственный за производство этого белка, является самым большим из всех известных генов, поэтому для проведения генной терапии ученые создали миниатюрную версию этого гена. Лучшими проводниками гена к мышцам ученые признали аденовирусы. Поэтому внутрь аденовируса они поместили нужный ген и ввели его мышам, страдающим от недостатка дистрофина. Результаты опыта оказались обнадеживающими. В других сходных работах носителями этого гена являются липосомы, микросферы, лактоферрин. Оригинальный подход к генотерапевтическому лечению МДД разрабатывается в Оксфордском университете группой под руководством Кей Девис (Kay Davies). Суть метода заключается в попытке дерепрессии аутосомного гомолога дистрофина – гена утрофина, продукт экспрессии которого мог бы быть способен компенсировать недостаток дистрофина во всех группах мышц. В эмбриогенезе человека приблизительно до семи недель развития дистрофин не экспрессируется и его функцию в мышцах выполняет белок утрофин. В промежутке между седьмой и 19 неделями развития экспрессируются оба белка и после 19 недели происходит замещение мышечного утрофина на дистрофин. После 19 недели эмбрионального развития утрофин обнаруживается только в области нервно-мышечных контактов. Белок утрофин, имея аутосомную локализацию разительно напоминает дистрофин своими N- и С- концевыми доменами, играющими решающую роль в функции дистрофина. Результаты экспериментов указывают на принципиальную возможность коррекции дефектов в мышечных волокнах лишенных дистрофина с помощью утрофина Установлено, что два препарата (L-arginine и heregulin) увеличивают производство белка атрофин в мышечных клетках мышей. Повышенное количество атрофина, вероятно, частично скомпенсирует отсутствие или недостаток белка дистрофин, который наблюдается при различных видах мышечной дистрофии. Перед тем, как эти препараты будут использоваться в лечении людей, ученым еще предстоит исследовать их безопасность и эффективность. В организме человека есть белок миостатин (myostatin), который ограничивает мышечный рост. Исследователи отметили улучшение состояния мышц мышей, больных мышечной дистрофией Дюшена, после блокировки этого белка. Биотехнологическая компания работает над созданием препарата, который сможет блокировать миостатин у мышей, и планирует дальнейшие тесты, которые позволили бы использовать эту технологию при лечении различных видов мышечной дистрофии у людей.
Мышечная дистрофия у детей относится к заболеваниям наследственного характера. Происходит нарушение функции волокон. Данная патология может передаваться по наследству. Только поддерживающая терапия может значительно улучшить состояние ребенка. Терапия заключается в том что, назначаются физиотерапевтические процедуры.
Если изучить все виды дистрофии, то их существует огромное множество. Но все они встречаются довольно редко. Наблюдается четыре типа дистрофий:
- Миопатия псевдогипертрофического происхождения;
- Болезнь Беккера;
- Миотония врожденного генеза;
- Дистрофия мышц плеча и дегенерация газового участка.
Самым распространенным среди всех дистрофий является миопатия псевдогипертрофического происхождения. Часто встречается у мальчиков, у девочек такая патология не диагностируется. По статистическим данным встречается у каждого трехтысячного ребенка. Первые признаки заболевания появляются в раннем детстве. Далее происходит прогрессирование снижения функции мышечных волокон, что приводит к падению активности.
Что касается Болезни Беккера, то встречается реже, чем предыдущая патология. Клинические проявления более скудные, их поначалу даже сложно диагностировать. Но, так или иначе, ребенок становится инвалидом.

Если диагностирована миотония дистрофического или врожденного происхождения, то в первую очередь ребенку тяжело дышать – это является основополагающим симптомом. После чего, сразу начинают ослабевать все группы мышц больного. Одинаково болеют как девочки, так и мальчики.
Среди всех дистрофий, самым редко встречающимся видом является поражение мышц плеча и газового пояса. Очень тяжелая патология, ухудшается качество жизни малыша.
Причины
Если мальчик заболевает миодистрофией, то исход крайне неблагоприятный. Такие больные живут максимум до 22-х лет. Если же у ребенка диагностирована болезнь Беккера, то исходом является инвалидность. Если проходит 20 лет с начала заболевания, то резко ухудшается активность человека, вплоть до прикованности к креслу.
Что касается миотонии врожденного генеза, то такие детки долго не живут. Но были случаи, когда новорожденные переживали первые сутки, далее могли прожить еще лет 15, но не более.
Абсолютно все разновидности патологии возникают из-за некоторых сбоев в генетической цепи. Если глубоко вдаться в подробности, нарушается структура на Х-хромосоме. Данная единица отвечает за выработку такого белка, как дистрофин. Он необходим для формирования нормальной функции мышечной ткани. Если происходит сбой в этом белке, то наступает дисфункция волокон и всего связочного аппарата организма.
Женский пол при данном заболевании является «переносчиком» патологического гена. Очень редко болеют девочки. Это связано с тем, что женский пол имеет две Х- хромосомы. Исходя из этого, происходит компенсация со второй Х-хромосомы.
Как только передается дефектный ген плоду мужского рода, то мальчик начинает заболевать. Это потому, что у мужского пола одна Х-хромосома. Поэтому компенсации от второй хромосомы не получится никогда.
Если сыновья являются непосредственными носителями патологического гена, то шансы передать по наследству становится около 50%. И около 50% всех девочек являются носителями мышечной дистрофии. Наблюдались казуистические случаи, когда ребенок заболевал, но в роду данной патологии не наблюдалось.
Диагностика
Выявить у детей раннего возраста заболевание не представляет никакой сложности. Достаточно изучить анамнез больного ребенка и провести клиническое обследование. Для точности доктор берет кровь больного и изучает ее в лабораторных условиях. Если наблюдается повышенное количество в крови креатинфосфокиназы, то можно подозревать, что ребенок болен. В нормальном состоянии этот фермент находится в мышечных волокнах пациента.
Также для проведения диагностики применяют:
- Электромиографию (с точностью выявляет активность электрического потенциала мышечной ткани);
- Эхокардиограмму (для исключения сердечной патологии, ведь сердце – это мышца);
- Биопсию мышечных волокон.

Биопсия берется у ребенка для того, чтобы изучить структурные изменения в волокнах. Это могут быть снижение коллагена или наличие избыточного отложение тканей жирового происхождения.
Лечение
В нынешнее время полностью купировать заболевание невозможно. Не существует каких-либо лекарственных препаратов или иных процедур, чтобы восстановить пораженные участки волокон.
Лечебная терапия данного патологического процесса направлена на приостановление прогрессирования деструкции. Для этого назначается:
- Препараты АТФ;
- Кортикостероиды;
- Лечебная физкультура;
- Предупреждение развития сколиоза, а также контрактуры ног.
Если выполнять данные пункты, то можно притормозить развитие заболевания. Проводить лечение нужно исключительно после рекомендации врача. Если не соблюдать все настояния специалиста или вовсе не лечить ребенка, то может настигнуть летальный исход.
- Советуем почитать про:
Профилактика
Для того чтобы предупредить данное заболевание у будущего потомства, существуют некоторые рекомендации. К ним относится:
- Если мать планирует забеременеть, то необходимо провести лабораторное исследование на присутствие в организме генов патологического происхождения. Также нужно тщательно изучить генеалогическое древо, чтобы исключить мышечную дистрофию.
- Обследовать отца на наличие патологических генов. Ведь это также играет важную роль для предотвращения рождения больного ребенка.
- Строгое соблюдение всех профилактических мер в случае осложнений у пациента.
Если соблюдать профилактические пункты, то можно исключить появление детей с данным заболеванием.
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.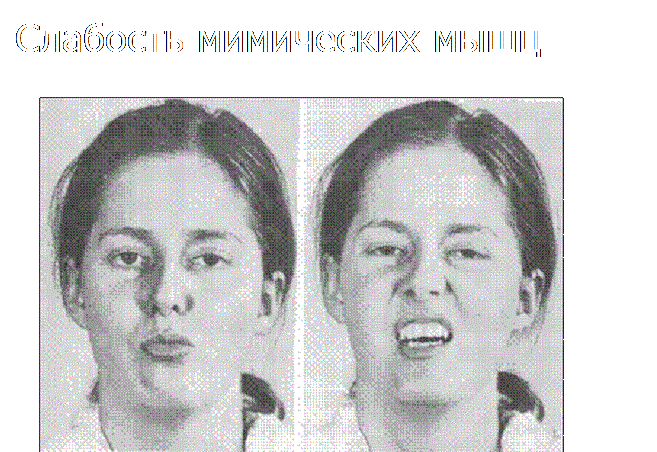
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!




 на тему")
