Distrofias musculares. Distrofia muscular em crianças: sintomas, tratamento, diagnóstico, causas
A distrofia muscular de Duchenne é uma doença genética associada a um distúrbio na estrutura das fibras musculares. As fibras musculares nesta doença eventualmente se rompem e a capacidade de movimento é perdida. A distrofia muscular de Duchenne é transmitida de maneira ligada ao gênero e afeta homens. Ela se manifesta já na infância. Além dos distúrbios musculares, a doença leva a deformidades esqueléticas e pode ser acompanhada de insuficiência respiratória e cardíaca, distúrbios mentais e endócrinos. Ainda não existe um tratamento radical para erradicar a doença. Todas as medidas existentes são apenas sintomáticas. É muito raro que os pacientes sobrevivam além dos 30 anos. Este artigo enfoca as causas, sintomas, diagnóstico e tratamento da distrofia muscular de Duchenne.
A doença foi descrita pela primeira vez em 1861 (segundo outras fontes - 1868) por um neurologista francês e leva seu nome. Não é tão raro: 1 caso em 3.500 recém-nascidos. De todas as conhecidas pela medicina, a distrofia muscular é a mais comum.

A distrofia muscular de Duchenne é baseada em um defeito genético do cromossomo X sexual.
Uma das seções do cromossomo X contém um gene que codifica a produção no corpo de uma proteína muscular especial chamada distrofina. A proteína distrofina forma a base das fibras musculares (miofibrilas) em nível microscópico. A função da distrofina é manter o esqueleto celular e garantir a capacidade das miofibrilas de sofrerem repetidos atos de contração e relaxamento. Na distrofia muscular de Duchenne, esta proteína está totalmente ausente ou é sintetizada de forma deficiente. O nível de distrofina normal não excede 3%. Isso leva à destruição das fibras musculares. Os músculos degeneram e são substituídos por gordura e tecido conjuntivo. Naturalmente, neste caso, o componente motor da atividade humana é perdido.
A doença é herdada de forma recessiva ligada ao cromossomo X. O que isto significa? Como todos os genes humanos são pareados, ou seja, duplicam-se, para que surjam alterações patológicas no corpo devido a uma doença hereditária, é necessário que surja um defeito genético em um cromossomo ou em seções semelhantes de ambos os cromossomos. Se a doença ocorre apenas com mutações em ambos os cromossomos, esse tipo de herança é denominado recessivo. Quando uma anomalia genética é detectada em apenas um cromossomo, mas a doença ainda se desenvolve, esse tipo de herança é chamado de dominante. O tipo recessivo só é possível quando cromossomos idênticos são afetados simultaneamente. Se o segundo cromossomo for “saudável”, a doença não ocorrerá. É por isso que a distrofia muscular de Duchenne é o destino dos homens, porque eles têm um cromossomo X em seu conjunto genético e o segundo cromossomo Y (pareado). Se um menino encontrar um cromossomo X “quebrado”, ele definitivamente desenvolverá o doença, porque o cromossomo saudável ele simplesmente não tem. Para que a distrofia muscular de Duchenne ocorra em uma menina, deve haver uma coincidência em seu genótipo de dois cromossomos X patológicos, o que é praticamente improvável (neste caso, o pai da menina deve estar doente e a mãe deve ter um X defeituoso cromossomo em sua composição genética). As meninas atuam apenas como portadoras da doença e a transmitem aos filhos. É claro que alguns casos da doença não são hereditários, mas ocorrem esporadicamente. Isto significa que uma mutação aparece espontaneamente na composição genética da criança. Uma mutação recém-surgida pode ser herdada (desde que a capacidade de reprodução seja preservada).
Sintomas da doença
A distrofia muscular de Duchenne sempre se manifesta antes dos 5 anos de idade. Na maioria das vezes, os primeiros sintomas aparecem antes dos 3 anos de idade. Todas as manifestações patológicas da doença podem ser divididas em vários grupos (dependendo da natureza das alterações):
- danos aos músculos esqueléticos;
- deformidades esqueléticas;
- danos ao músculo cardíaco;
- Deficiência mental;
- distúrbios endócrinos.
Dano muscular esquelético

Os danos ao tecido muscular são a principal manifestação da doença. Causa fraqueza muscular generalizada. Os sintomas iniciais passam despercebidos.
As crianças nascem sem quaisquer desvios especiais. No entanto, seu desenvolvimento motor fica aquém do de seus pares. Essas crianças são menos ativas e móveis em termos de atividade motora. Embora a criança seja muito pequena, isso está frequentemente associado a características temperamentais e nenhuma atenção é dada às mudanças iniciais.
Sinais óbvios aparecem assim que a caminhada começa. As crianças muitas vezes caem e andam na ponta dos pés. Ressalta-se que essas violações não são interpretadas durante os primeiros passos da criança, pois a marcha ereta está inicialmente associada a quedas e falta de jeito para todas as crianças. Embora a maioria de seus colegas consiga andar com bastante confiança, os meninos com distrofia muscular de Duchenne continuam a cair teimosamente.
Quando a criança aprende a falar, começa a reclamar de fraqueza e cansaço, além de intolerância à atividade física. Correr, escalar, saltar e outras atividades favoritas das crianças não são atraentes para uma criança com distrofia muscular de Duchenne.
O andar dessas crianças lembra o de um pato: elas parecem balançar de um pé para o outro.
Uma manifestação peculiar da doença é o sintoma de Govers. É o seguinte: quando uma criança tenta se levantar dos joelhos, agachar-se ou cair no chão, ela usa as mãos para ajudar os músculos fracos das pernas. Para isso, ele apoia as mãos sobre si mesmo, “subindo a escada, sozinho”.
A distrofia muscular de Duchenne apresenta um padrão ascendente de fraqueza muscular. Isto significa que a fraqueza aparece primeiro nas pernas, depois se espalha para a pélvis e tronco, depois para os ombros, pescoço e finalmente para os braços, músculos respiratórios e cabeça.
Apesar do fato de que, com esta doença, as fibras musculares são destruídas e a atrofia se desenvolve, externamente alguns músculos podem parecer bastante normais ou até mesmo aumentados. Desenvolve-se a chamada pseudo-hipertrofia dos músculos. Na maioria das vezes, esse processo é perceptível na panturrilha, nos músculos glúteos e deltóides e nos músculos da língua. O lugar das fibras musculares desintegradas é ocupado pelo tecido adiposo, por isso se cria o efeito de um bom desenvolvimento muscular que, quando testado, revela-se completamente errado.
O processo atrófico nos músculos é sempre simétrico. A direção ascendente do processo leva ao aparecimento de cintura de “vespa”, omoplatas “em forma de asa” (as omoplatas ficam atrás do corpo, como asas) e o sintoma de “cinturas escapulares soltas” (quando a cabeça parece cair nos ombros ao tentar levantar a criança pelas axilas). O rosto fica hipomímico, os lábios podem engrossar (substituição dos músculos por tecido adiposo e conjuntivo). A pseudohipertrofia da língua causa distúrbios da fala.
A destruição dos músculos é acompanhada pelo desenvolvimento de contraturas musculares e encurtamento dos tendões (claramente perceptível no exemplo do tendão de Aquiles).
Os reflexos do tendão (joelho, Aquiles, bíceps, tríceps, etc.) diminuem gradualmente. Os músculos são firmes ao toque, mas indolores. O tônus muscular geralmente diminui.
A progressão gradual da fraqueza muscular leva ao fato de que, aos 10-12 anos de idade, muitas crianças perdem a capacidade de se mover de forma independente e precisam de uma cadeira de rodas. A capacidade de ficar em pé dura, em média, até os 16 anos.
Separadamente, deve-se dizer sobre o envolvimento dos músculos respiratórios no processo patológico. Isso é observado após a adolescência. A fraqueza do diafragma e de outros músculos envolvidos no ato respiratório leva a uma diminuição gradual da capacidade vital dos pulmões e dos volumes de ventilação. Isso é especialmente perceptível à noite (aparecem iscas de asfixia), então as crianças podem ter medo antes de dormir. Desenvolve-se insuficiência respiratória, o que agrava o curso de infecções intercorrentes.
Deformidades esqueléticas

Estes são sintomas que acompanham as alterações musculares. As crianças desenvolvem gradualmente uma curva lombar aumentada (lordose), uma curvatura lateral da coluna torácica (escoliose) e uma postura curvada (cifose), e a forma do pé muda. Com o tempo, desenvolve-se osteoporose difusa. Esses sintomas pioram ainda mais os distúrbios do movimento.
Danos ao músculo cardíaco
É um sintoma obrigatório da distrofia muscular de Duchenne. Os pacientes desenvolvem cardiomiopatia (hipertrófica ou dilatada). Clinicamente, isso se manifesta como distúrbios do ritmo cardíaco e alterações na pressão arterial. Os limites do coração aumentam, mas um coração tão grande tem pouca funcionalidade. Eventualmente, desenvolve-se insuficiência cardíaca. A combinação de insuficiência cardíaca grave com distúrbios respiratórios no contexto de uma infecção associada pode ser a causa de morte em pacientes com distrofia muscular de Duchenne.
Deficiência mental
Isto não é obrigatório, mas é um possível sinal da doença. Está associada à deficiência de uma forma especial de distrofina, a apodistrofina, encontrada no cérebro. As deficiências intelectuais variam de leves a idiotas. Além disso, a gravidade da deficiência mental não está de forma alguma relacionada com o grau dos distúrbios musculares. A desadaptação social pela impossibilidade de circular livremente e frequentar instituições de acolhimento de crianças (creches, escolas) contribui para o agravamento dos distúrbios cognitivos.
Distúrbios endócrinos
Ocorre em 30-50% dos pacientes. Podem ser bastante variados, mas na maioria das vezes é obesidade com deposição predominante de gordura na região das glândulas mamárias, coxas, nádegas, cintura escapular, subdesenvolvimento (ou disfunção) dos órgãos genitais. Os pacientes geralmente são de baixa estatura.
A distrofia muscular de Duchenne está progredindo constantemente. Aos 15-20 anos, quase todos os pacientes são incapazes de cuidar de si próprios devido à imobilidade. No final, somam-se as infecções bacterianas (órgãos respiratórios e urinários, escaras infectadas com cuidados insuficientes) que, num contexto de insuficiência cardíaca e respiratória, levam à morte. Poucos pacientes sobrevivem aos 30 anos.
Diagnóstico

O diagnóstico da distrofia muscular de Duchenne é baseado em vários tipos de estudos, sendo o principal deles um teste genético (diagnóstico de DNA).
Somente a detecção de um defeito no cromossomo X na região responsável pela síntese da distrofina confirma o diagnóstico de forma confiável. Antes de tal análise ser realizada, o diagnóstico é preliminar.
Outros métodos de pesquisa podem ser usados:
- determinação da atividade da creatina fosfoquinase (CPK). Esta enzima reflete a morte das fibras musculares. Sua concentração na distrofia muscular de Duchenne excede a norma dezenas e centenas de vezes antes dos 5 anos de idade. Mais tarde, o nível da enzima diminui gradualmente porque algumas fibras musculares já estão irreversivelmente destruídas;
- eletromiografia. Este método permite confirmar o fato de que a doença se baseia em alterações musculares primárias e os condutores nervosos estão completamente intactos;
- biópsia muscular. É usado para determinar o conteúdo da proteína distrofina no músculo. No entanto, devido às melhorias no diagnóstico genético nas últimas décadas, este procedimento traumático ficou em segundo plano;
- testes respiratórios (estudo da capacidade vital dos pulmões), ECG, ultrassonografia do coração. Esses métodos não são utilizados para estabelecer um diagnóstico, mas são necessários para identificar alterações patológicas nos sistemas respiratório e cardiovascular, a fim de corrigir distúrbios existentes.
A identificação de uma criança doente na família significa que o genótipo da mãe contém um cromossomo X patológico. Em casos raros, a mãe pode ser saudável se a mutação ocorreu por acaso na criança. Ter um cromossomo X defeituoso acarreta um risco para gestações subsequentes. Portanto, essas famílias devem ser aconselhadas por um geneticista. Quando ocorrem gestações repetidas, é oferecido aos pais o diagnóstico pré-natal, ou seja, um estudo do genótipo do feto para excluir doenças hereditárias, incluindo a distrofia muscular de Duchenne.
Para o estudo, você precisará de células fetais obtidas por meio de vários procedimentos em diferentes estágios da gravidez (por exemplo, amostragem de vilosidades coriônicas, amniocentese e outros). E embora esses procedimentos médicos representem um certo risco para a gravidez, eles podem responder com precisão à pergunta: o feto tem uma doença genética?
Tratamento
A distrofia muscular de Duchenne é atualmente uma doença incurável. Você pode ajudar uma criança (adulto) a prolongar o tempo de atividade física utilizando diversos métodos para manter a força muscular, compensando alterações nos sistemas cardiovascular e respiratório.
Apesar disso, as previsões dos cientistas para a cura completa desta doença são bastante otimistas, uma vez que os primeiros passos nesse sentido já foram dados.
Atualmente, os medicamentos usados para tratar a distrofia muscular de Duchenne incluem:
- esteróides (com uso regular podem reduzir a fraqueza muscular);
- Agonistas β-2-adrenérgicos (também aumentam temporariamente a força muscular, mas não retardam a progressão da doença).
O uso de agonistas β-2-adrenérgicos (Albuterol, Formoterol) não possui reconhecimento estatisticamente confiável, pois há pouca experiência com seu uso nesta patologia. As alterações no estado de saúde de um grupo de pacientes em uso desses medicamentos foram monitoradas durante um ano. Portanto, não é possível afirmar que funcionam por mais tempo.
A base do tratamento hoje são os esteróides. Acredita-se que seu uso permite manter a força muscular por algum tempo, ou seja, podem retardar a progressão da doença. Além disso, foi demonstrado que os esteróides reduzem o risco de escoliose na distrofia muscular de Duchenne. Mesmo assim, as capacidades destes medicamentos são limitadas e a doença irá progredir de forma constante.
 Quando começa o tratamento hormonal? Acredita-se que o momento ideal para iniciar a terapia é a fase da doença em que as habilidades motoras não melhoram, mas ainda não estão piorando. Isso geralmente acontece entre as idades de 4 e 6 anos. Os medicamentos mais utilizados são Prednisolona e Deflazacort. As doses são prescritas individualmente. Os medicamentos são utilizados enquanto houver efeito clínico visível. Quando se inicia a fase de progressão da doença, a necessidade do uso de esteroides desaparece, sendo gradativamente (!) abolidos.
Quando começa o tratamento hormonal? Acredita-se que o momento ideal para iniciar a terapia é a fase da doença em que as habilidades motoras não melhoram, mas ainda não estão piorando. Isso geralmente acontece entre as idades de 4 e 6 anos. Os medicamentos mais utilizados são Prednisolona e Deflazacort. As doses são prescritas individualmente. Os medicamentos são utilizados enquanto houver efeito clínico visível. Quando se inicia a fase de progressão da doença, a necessidade do uso de esteroides desaparece, sendo gradativamente (!) abolidos.
Entre os medicamentos, os medicamentos cardíacos (antiarrítmicos, metabólicos, inibidores da enzima conversora de angiotensina) também são utilizados para a distrofia muscular de Duchenne. Eles permitem combater os aspectos cardíacos da doença.
Entre os métodos de tratamento não medicamentoso, a fisioterapia e os cuidados ortopédicos desempenham um papel significativo. As técnicas fisioterapêuticas permitem manter a flexibilidade e mobilidade das articulações por mais tempo do que sem seu uso, além de manter a força muscular. Está comprovado que a atividade física moderada tem um efeito benéfico no curso da doença, mas a inatividade e o repouso no leito, pelo contrário, contribuem para uma progressão ainda mais rápida da doença. Portanto, é necessário manter a atividade física viável pelo maior tempo possível, mesmo após o paciente ter “mudado” para a cadeira de rodas. São indicados cursos regulares de massagem. A natação tem um efeito positivo no bem-estar do paciente.
Os dispositivos ortopédicos podem facilitar significativamente a vida do paciente. A sua lista é bastante ampla e variada: incluem vários tipos de verticalizadores (ajudam a manter a posição em pé), e dispositivos para se levantar de forma independente, e cadeiras de rodas eléctricas, e talas especiais para eliminar contraturas na parte inferior da perna (usadas mesmo à noite) e espartilhos para a coluna e talas longas para as pernas (órteses joelho-tornozelo) e muito mais.
Quando a doença atinge a musculatura respiratória e a respiração espontânea torna-se ineficaz, é possível utilizar dispositivos de ventilação pulmonar artificial de diversas modificações.
E, no entanto, mesmo a utilização combinada de todas estas medidas não nos permite superar a doença. Hoje, há uma série de áreas de pesquisa promissoras que podem se tornar um avanço no tratamento da distrofia muscular de Duchenne. Os mais comuns entre eles incluem:
- terapia genética (introdução do gene “correto” usando partículas virais, entrega de construções genéticas como parte de lipossomas, oligopeptídeos, transportadores poliméricos, etc.);
- regeneração de fibras musculares utilizando células-tronco;
- transplante de células miogênicas capazes de sintetizar distrofina normal;
- salto de exon (usando oligorribonucleotídeos antisense) como uma tentativa de retardar a progressão da doença e mitigar seu curso;
- substituição da distrofina por outra proteína, a utrofina, cujo gene foi decifrado. A técnica foi testada em ratos e deu resultados positivos.
Cada um dos novos desenvolvimentos traz esperança para os pacientes com distrofia muscular de Duchenne de uma recuperação completa.
Assim, a distrofia muscular de Duchenne é um problema genético nos homens. A doença é caracterizada por fraqueza muscular progressiva devido à destruição das fibras musculares. Atualmente, é uma doença incurável, mas muitos cientistas ao redor do mundo estão trabalhando para criar uma forma radical de combatê-la.
Filme de animação "Distrofia muscular de Duchenne", inglês. narração, legendas em russo:
A distrofia muscular é na verdade um grupo de doenças hereditárias caracterizadas por atrofia simétrica progressiva dos músculos esqueléticos, ocorrendo sem dor e perda de sensibilidade nos membros. Paradoxalmente, os músculos afetados podem aumentar de tamanho devido ao crescimento de tecido conjuntivo e depósitos de gordura, dando a falsa impressão de músculos fortes.
Ainda não há cura para a distrofia muscular. Existem quatro tipos principais desta patologia. A distrofia muscular de Duchenne é a mais comum (50% de todos os casos). A doença geralmente começa na primeira infância e leva à morte aos 20 anos. A distrofia muscular de Becker se desenvolve mais lentamente, os pacientes vivem mais de 40 anos. As distrofias ombro-escápulo-faciais e de cinturas geralmente não afetam a expectativa de vida.
Causas
Desenvolvimento de distrofia muscular. causada por vários genes. A distrofia muscular de Duchenne e a distrofia muscular de Becker são causadas por genes no cromossomo sexual e afetam apenas homens. As distrofias escapuloumeral-faciais e membros-lombares não estão associadas aos cromossomos sexuais; Tanto homens como mulheres adoecem com eles.
Sintomas
Todos os tipos de distrofia muscular causam atrofia muscular progressiva.
Diagnóstico
O médico examina a criança, tira dúvidas sobre doenças dos familiares e prescreve alguns exames. Se um de seus parentes sofreu de distrofia muscular, o médico descobrirá como a distrofia progrediu. Ao analisar os dados obtidos, é possível prever o que espera a criança. Caso não existam pacientes com distrofia muscular na família, a eletromiografia avaliará o funcionamento dos nervos dos músculos afetados e determinará a presença de distrofia muscular; o exame de um pedaço de tecido muscular () pode mostrar alterações celulares e a presença de depósitos de gordura.
Em centros médicos equipados com os mais modernos equipamentos para a realização de pesquisas biológicas e imunológicas moleculares, eles podem determinar com precisão se uma criança sofrerá de distrofia muscular. Esses centros também podem rastrear pais e parentes quanto à presença de genes que determinam o desenvolvimento da distrofia muscular de Duchenne e da distrofia muscular de Becker.
Tipos de doença
De acordo com a gravidade da doença e o momento de seu início, distinguem-se:
A distonia de Duchenne manifesta-se em idade precoce (entre os 3 e os 5 anos). As crianças doentes gingam, têm dificuldade em subir escadas, caem frequentemente e não conseguem correr. Quando levantam os braços, as omoplatas “ficam atrás” do corpo - esse sintoma é chamado de “omoplatas aladas”. Normalmente, uma criança com distrofia muscular fica confinada a uma cadeira de rodas aos 9-12 anos de idade. A fraqueza progressiva do músculo cardíaco leva à morte por insuficiência cardíaca súbita, insuficiência respiratória ou infecção.
Embora a distrofia de Becker tenha muitas semelhanças com a distrofia de Duchenne, ela se desenvolve muito mais lentamente. Os sintomas aparecem por volta dos 5 anos de idade, mas depois dos 15 anos, as crianças afetadas geralmente ainda mantêm a capacidade de andar e, às vezes, muito mais tarde.
A distrofia humoescapulofacial se desenvolve lentamente e seu curso é relativamente benigno. Na maioria das vezes, a doença começa antes dos 10 anos, mas pode aparecer no início da adolescência. As crianças que posteriormente desenvolvem esta patologia sugam mal na infância; quando envelhecem, não conseguem franzir os lábios como um assobio ou levantar os braços acima da cabeça. Em crianças doentes, seus rostos são caracterizados pela inatividade ao rir ou chorar e, às vezes, são notadas expressões faciais diferentes do normal.
Ações do Paciente
Se você está preocupado com a possibilidade de seu filho estar desenvolvendo distrofia muscular, você pode trazer fotos ou vídeos que ilustrem seus problemas específicos. Traga um parente ou amigo que também ouvirá as informações fornecidas pelo médico.
Tratamento
Ainda não existe um remédio que possa impedir a progressão da atrofia muscular na distrofia muscular. No entanto, dispositivos ortopédicos, bem como terapia por exercícios, fisioterapia e cirurgia para corrigir contraturas, podem manter a mobilidade de uma criança ou adolescente por um tempo.
Os familiares com histórico de distrofia muscular devem procurar aconselhamento genético para determinar se existe risco de transmissão da doença a um futuro filho.
Complicações
Alguns tipos de distrofia muscular encurtam a vida de uma pessoa e muitas vezes afetam os músculos associados à respiração. Mesmo com a respiração mecânica melhorada, as pessoas que têm distrofia muscular de Duchenne – o tipo mais comum de distrofia muscular – normalmente morrem de insuficiência respiratória antes de atingirem os 40 anos.
Muitos tipos de distrofia muscular também podem reduzir a eficiência do músculo cardíaco. Se a doença afetar os músculos associados à deglutição, podem surgir problemas nutricionais.
À medida que a fraqueza muscular progride, a mobilidade torna-se um problema. Muitas pessoas que desenvolvem distrofia muscular podem precisar usar cadeira de rodas. No entanto, a imobilidade articular prolongada associada ao uso da cadeira de rodas pode piorar as contraturas nas quais os membros giram e travam em uma posição interna.
As contraturas também podem desempenhar um papel no desenvolvimento da escoliose, causando uma curvatura da coluna vertebral, o que reduz ainda mais a eficiência dos pulmões em pessoas com distrofia muscular.
Prevenção
Como as infecções respiratórias podem se tornar um problema nos estágios posteriores da distrofia muscular, é importante ser vacinado contra a pneumonia e também tomar vacinas regulares contra a gripe.
![]()
Descrição:
A distrofia muscular é um grupo de doenças hereditárias crônicas dos músculos esqueléticos humanos, manifestadas por fraqueza e degeneração muscular. Existem nove formas diferentes de distrofia muscular. Eles diferem em características como a idade em que a doença começa, a localização dos músculos afetados, a gravidade da fraqueza muscular, a taxa de progressão da distrofia e o tipo de sua herança. As duas formas mais comuns são a distrofia muscular de Duchenne e a distrofia muscular miotônica.
Sintomas:
Distrofia de Duchenne. Mutação recessiva do cromossomo X do gene da distrofina. Sinais clínicos: início antes dos 5 anos; fraqueza progressiva dos músculos da cintura pélvica e escapular; incapacidade de andar após os 12 anos; cifoescoliose; insuficiência respiratória aos 20-30 anos de idade. Envolvimento de outros sistemas orgânicos: ; diminuição da inteligência.
Distrofia de Becker. Mutação recessiva do cromossomo X do gene da distrofina. Características clínicas: início precoce ou tardio; fraqueza lentamente progressiva dos músculos da cintura pélvica e escapular; manter a capacidade de andar após os 15 anos; insuficiência respiratória após 40 anos. Envolvimento de outros sistemas orgânicos: cardiomiopatia.
Distrofia miotônica. Autossômico dominante; expansão da região instável do DNA do cromossomo 19ql3,3. Sinais clínicos: início em qualquer idade; fraqueza lentamente progressiva dos músculos das pálpebras, face, pescoço, músculos distais dos membros; miotonia. Envolvimento de outros sistemas orgânicos: distúrbios de condução cardíaca; Transtornos Mentais, Desordem Mental; , frontal; gônadas
Distrofia humoescapulofacial.
Autossômico dominante; frequentemente mutações do cromossomo 4q35. Sinais clínicos: início antes dos 20 anos; fraqueza muscular lentamente progressiva da região facial, cintura escapular e dorsiflexão do pé. Envolvimento de outros sistemas orgânicos: hipertensão; surdez.
Cintura escapular e pélvica (várias doenças são possíveis). Autossômico recessivo ou dominante. Características clínicas: início na primeira infância até a meia-idade; fraqueza lentamente progressiva dos músculos do ombro e da cintura pélvica. Envolvimento de outros sistemas orgânicos: cardiomiopatia.
Distrofia oculofaríngea. Autossômico dominante (Canadá francês ou Espanha). Sinais clínicos: início aos 50-60 anos; fraqueza muscular lentamente progressiva: pálpebras externas, pálpebras, face e faringe; acalasia cricofaríngea. Envolvimento de outros sistemas orgânicos: cerebral, ocular.
Distrofia congênita. Inclui diversas doenças, incluindo tipos de Fukuyama e displasia cerebroocular). Autossômica recessiva. Sinais clínicos: início ao nascimento; hipotensão, atraso no desenvolvimento; em alguns casos - insuficiência respiratória precoce, em outros - curso mais favorável da doença.
Causas:
A doença é causada por um gene autossômico dominante com expressividade bastante variável (o risco de transmissão para parentes de primeiro grau é de 50%). A doença é causada por amplificação, ou seja, um aumento no número de trigêmeos CTG em um determinado locus do cromossomo 19 (distrofia miotônica tipo 1) ou CCTG no cromossomo 3 (distrofia miotônica tipo 2). A distrofia miotônica tipo 2 é pouco estudada. Acredita-se que ocorra em apenas 2% dos casos (mas pode ser muito mais comum); não relacionado ao tipo 1; muito provavelmente não é a causa das formas congênitas de distrofia quando o portador é a mãe. Para o tipo 1, foi comprovado que o número de repetições de nucleotídeos aumenta à medida que a mutação é transmitida de geração em geração. A gravidade da doença está claramente correlacionada com o número destas repetições. O maior número deles é determinado na forma congênita grave da doença. O mecanismo identificado explica o fenômeno da antecipação - a gravidade e o início mais precoce da doença nas gerações descendentes. Por exemplo, se uma análise genética mostrou que um dos pais tem um certo número de repetições CTG, então seu filho terá um número ainda maior de repetições desse trigêmeo.
Tratamento:
Até o momento, não há como prevenir ou retardar a progressão desta doença. A terapia visa principalmente combater complicações, como a deformidade da coluna vertebral, que se desenvolve devido à fraqueza dos músculos das costas, ou a predisposição à pneumonia devido à fraqueza dos músculos respiratórios. Fenitoína, procainamida, quinina são utilizadas no tratamento da miotonia, mas é necessária cautela em pacientes com doenças cardíacas (risco de piora da condução cardíaca). O implante de marca-passo é necessário para pacientes com síncope ou bloqueio cardíaco. O medicamento fenigidina é recomendado para o tratamento de doenças cardíacas. O uso de aparelhos ortopédicos pode fortalecer os pés “caídos”, estabilizar as articulações do tornozelo e reduzir a frequência de quedas. Um treinamento bem escolhido também pode ter um efeito positivo no curso da doença. Na presença de atrofia, são utilizados esteróides anabolizantes (retabolil, nerobol) e terapia restauradora geral. Nos casos em que há sintomas miotônicos significativamente pronunciados, são prescritos cursos de difenina de 0,03-0,05 g 3 vezes ao dia, com duração de 2-3 semanas. Supõe-se que a difenina tenha um efeito inibitório na condução sináptica e reduza a atividade pós-tetânica nos músculos. Com o aumento da sonolência, que muitas vezes acompanha a distrofia miotônica, observa-se um efeito positivo ao tomar selegilina. Recomenda-se também tomar alguns suplementos alimentares: coenzima Q10 (100 mg/dia), vitamina E (200 UI/dia) e selênio (200 mcg/dia), lecitina (20 g/dia).
Uma cura eficaz para esta doença só é possível com a ajuda da terapia genética, que está agora a ser intensamente desenvolvida. Numerosos experimentos mostram melhorias na condição das fibras musculares no tratamento de algumas formas de distrofia muscular. Nas distrofias de Duchenne e Becker, há produção insuficiente da proteína muscular distrofina. O gene responsável pela produção desta proteína é o maior de todos os genes conhecidos, por isso os cientistas criaram uma versão em miniatura deste gene para realizar a terapia genética. Os cientistas reconheceram os adenovírus como os melhores condutores de genes para os músculos. Portanto, eles colocaram o gene desejado dentro do adenovírus e o injetaram em ratos que sofriam de deficiência de distrofina. Os resultados do experimento foram encorajadores. Em outros trabalhos semelhantes, lipossomas, microesferas e lactoferrina são portadores desse gene. Uma abordagem original à terapia genética para DMD está sendo desenvolvida na Universidade de Oxford por um grupo liderado por Kay Davies. A essência do método é uma tentativa de desreprimir o homólogo autossômico da distrofina - o gene da utrofina, cujo produto de expressão poderia compensar a falta de distrofina em todos os grupos musculares. Na embriogênese humana, a distrofina não é expressa até aproximadamente sete semanas de desenvolvimento, e sua função nos músculos é desempenhada pela proteína utrofina. Entre a sétima e a 19ª semanas de desenvolvimento, ambas as proteínas são expressas e após a 19ª semana a utrofina muscular é substituída pela distrofina. Após a 19ª semana de desenvolvimento embrionário, a utrofina é encontrada apenas na área de contatos neuromusculares. A proteína utrofina, tendo localização autossômica, assemelha-se notavelmente à distrofina com seus domínios N e C-terminais, que desempenham um papel decisivo na função da distrofina. Os resultados experimentais indicam a possibilidade fundamental de corrigir defeitos nas fibras musculares carentes de distrofina com o auxílio da utrofina.Foi estabelecido que dois medicamentos (L-arginina e heregulina) aumentam a produção da proteína utrofina nas células musculares de camundongos. É provável que o aumento da quantidade de utrofina compense parcialmente a ausência ou deficiência da proteína distrofina que é observada em vários tipos de distrofia muscular. Antes que esses medicamentos sejam usados em humanos, os cientistas ainda precisam estudar sua segurança e eficácia. O corpo humano contém uma proteína chamada miostatina, que limita o crescimento muscular. Os pesquisadores notaram melhorias na saúde muscular de ratos com distrofia muscular de Duchenne após o bloqueio desta proteína. A empresa de biotecnologia está trabalhando para desenvolver um medicamento que possa bloquear a miostatina em ratos e planeja mais testes que possam usar a tecnologia para tratar vários tipos de distrofia muscular em humanos.
A distrofia muscular em crianças é uma doença hereditária. A função da fibra está prejudicada. Esta patologia pode ser herdada. Somente a terapia de suporte pode melhorar significativamente a condição da criança. A terapia consiste na prescrição de procedimentos fisioterapêuticos.
Se você estudar todos os tipos de distrofia, verá que há um grande número deles. Mas todos eles são bastante raros. Existem quatro tipos de distrofias:
- Miopatia de origem pseudo-hipertrófica;
- doença de Becker;
- Miotonia de origem congênita;
- Distrofia dos músculos do ombro e degeneração da área gasosa.
A mais comum entre todas as distrofias é a miopatia de origem pseudo-hipertrófica. Frequentemente encontrada em meninos, esta patologia não é diagnosticada em meninas. Segundo as estatísticas, ocorre a cada três mil crianças. Os primeiros sinais da doença aparecem na primeira infância. Em seguida, progride o declínio da função das fibras musculares, o que leva a uma diminuição da atividade.
Quanto à doença de Becker, é menos comum que a patologia anterior. As manifestações clínicas são mais esparsas e até difíceis de diagnosticar num primeiro momento. Mas, de uma forma ou de outra, a criança fica deficiente.

Se for diagnosticada miotonia de origem distrófica ou congênita, primeiro a criança tem dificuldade para respirar - este é um sintoma fundamental. Depois disso, todos os grupos musculares do paciente começam imediatamente a enfraquecer. Tanto as meninas como os meninos adoecem igualmente.
Entre todas as distrofias, o tipo mais raro são as lesões nos músculos do ombro e do trato gastrointestinal. Patologia muito grave, piora a qualidade de vida do bebê.
Causas
Se um menino desenvolver distrofia muscular, o resultado será extremamente desfavorável. Esses pacientes vivem no máximo 22 anos. Se uma criança for diagnosticada com doença de Becker, o resultado é incapacidade. Se passarem 20 anos desde o início da doença, a atividade da pessoa deteriora-se acentuadamente, até ao ponto de ficar confinada a uma cadeira.
Quanto à miotonia de origem congênita, essas crianças não vivem muito. Mas houve casos em que os recém-nascidos sobreviveram ao primeiro dia, depois puderam viver mais 15 anos, mas não mais.
Absolutamente todos os tipos de patologia surgem devido a algumas falhas na cadeia genética. Se você entrar em detalhes, a estrutura do cromossomo X será interrompida. Esta unidade é responsável pela produção de uma proteína como a distrofina. É necessário para a formação da função normal do tecido muscular. Se houver um mau funcionamento dessa proteína, ocorre disfunção das fibras e de todo o aparelho ligamentar do corpo.
Nesta doença, o sexo feminino é o “portador” do gene patológico. As meninas raramente ficam doentes. Isso se deve ao fato do sexo feminino possuir dois cromossomos X. Com base nisso, a compensação ocorre a partir do segundo cromossomo X.
Assim que o gene defeituoso é transmitido a um feto masculino, o menino começa a adoecer. Isso ocorre porque os homens têm um cromossomo X. Portanto, a compensação a partir do segundo cromossomo nunca será possível.
Se os filhos forem portadores diretos do gene patológico, as chances de transmiti-lo passam a ser de cerca de 50%. E cerca de 50% de todas as meninas são portadoras de distrofia muscular. Houve casos ocasionais em que uma criança adoeceu, mas esta patologia não foi observada na família.
Diagnóstico
Não é difícil detectar a doença em crianças pequenas. Basta estudar o histórico médico da criança doente e realizar um exame clínico. Para maior precisão, o médico coleta o sangue do paciente e o estuda em laboratório. Se houver uma quantidade aumentada de creatinofosfoquinase no sangue, você pode suspeitar que a criança está doente. Normalmente, esta enzima é encontrada nas fibras musculares do paciente.
Também usado para diagnóstico:
- Eletromiografia (detecta com precisão a atividade do potencial elétrico do tecido muscular);
- Ecocardiograma (para excluir patologia cardíaca, pois o coração é um músculo);
- Biópsia de fibra muscular.

Uma biópsia é feita na criança para estudar alterações estruturais nas fibras. Isso pode ser uma diminuição do colágeno ou a presença de deposição excessiva de tecido de origem gordurosa.
Tratamento
Atualmente, é impossível parar completamente a doença. Não existem medicamentos ou outros tratamentos para restaurar as áreas afetadas das fibras.
A terapia terapêutica para esse processo patológico visa interromper a progressão da destruição. Para tanto é prescrito:
- Preparações de ATP;
- Corticosteróides;
- Fisioterapia;
- Prevenção do desenvolvimento de escoliose, bem como contraturas nas pernas.
Se você seguir essas etapas, poderá retardar o desenvolvimento da doença. O tratamento deve ser realizado somente após recomendação de um médico. Se você não seguir todas as insistências do especialista ou não tratar a criança de jeito nenhum, pode resultar em morte.
- Recomendamos ler sobre:
Prevenção
Para prevenir esta doença nos futuros descendentes, existem algumas recomendações. Esses incluem:
- Se a mãe planeja engravidar, é necessário realizar um exame laboratorial para determinar a presença de genes de origem patológica no organismo. Você também precisa estudar cuidadosamente a árvore genealógica para descartar a distrofia muscular.
- Examine o pai quanto à presença de genes patológicos. Afinal, isso também desempenha um papel importante na prevenção do nascimento de uma criança doente.
- Cumprimento estrito de todas as medidas preventivas em caso de complicações no paciente.
Se você seguir os pontos preventivos, poderá excluir o aparecimento de crianças com esta doença.
Na neurologia moderna, existe um grande número de doenças cuja natureza não pode ser explicada racionalmente pelos especialistas. Essas doenças incluem um grupo de doenças como a distrofia muscular. Existem nove variedades desta doença no total, mas primeiro o mais importante…
A distrofia muscular é uma doença hereditária crônica que resulta em danos ao sistema muscular humano. Os músculos afetados deixam de funcionar normalmente, tornam-se mais finos e, em sua localização no corpo, uma camada de gordura aumenta gradualmente.
Tipos de distrofia muscular
Na neurologia moderna, esta doença é classificada em nove doenças diferentes. A classificação da doença está relacionada a:
- localização de distúrbios musculares;
- características da doença;
- agressividade do desenvolvimento;
- idade.
Então, a distrofia muscular acontece:
- Duchenne;
- miotônico (doença de Steinert);
- Becker;
- Erba Rota;
- forma juvenil de distrofia de Erb-Roth;
- ombro - formato facial escapular (Landouzy-Dejerine);
- miopatia alcoólica;
- forma distal;
- Distrofia muscular de Emery-Dreyfus.
Distrofia de Duchenne
A forma mais conhecida é a distrofia progressiva de Duchenne (distrofia pseudo-hipertrófica, merosina - distrofia congênita negativa). Esse tipo de doença se manifesta na infância, dos dois aos cinco anos. Em primeiro lugar, esta doença afeta os músculos das extremidades inferiores, que, apesar do sedentarismo em pacientes jovens, aumentam gradualmente de tamanho. Esse recurso está associado ao crescimento de tecido adiposo em vez de músculo.

À esquerda está uma criança saudável, à direita está uma criança doente
Gradualmente, à medida que a doença progride, ela se desloca para a parte superior e afeta os músculos dos membros superiores. Via de regra, aos doze anos o pequeno paciente para de se movimentar completamente. A taxa de mortalidade desta doença é muito alta; aproximadamente 85-90% dos pacientes não sobrevivem até os 20 anos.
O grupo de risco é o sexo masculino, pois a doença praticamente não atinge as meninas.
Doença de Steinert
Não é à toa que a distrofia congênita de Steiner é chamada de miotônica, pois como resultado da progressão da doença, o relaxamento muscular ocorre muito lentamente após a contração (esse fenômeno é chamado de miotonia).
Esta doença, ao contrário da anterior, é comum em adultos entre 20 e 40 anos. Existem também casos de progressão da doença em crianças, geralmente na infância, no entanto, esta é a excepção e não a regra.

Sinais faciais de miotonia (boca e pálpebras abertas)
A doença não depende do género e afecta igualmente homens e mulheres. Observa-se que a doença se manifesta como fraqueza dos músculos faciais, bem como dos membros. A progressão, ao contrário da doença de Duchenne, ocorre lentamente.
Uma característica distintiva da doença é a possibilidade de lesões não só nos músculos dos membros, mas também nos músculos internos (músculo cardíaco), o que por sua vez representa um grande perigo para a vida humana.
Doença de Becker
Essa forma da doença também progride por muito tempo e o paciente se sente bem por muito tempo. Uma exacerbação da doença pode ocorrer no contexto de lesões ou de várias doenças do sistema nervoso, que no seu curso acelerarão o desenvolvimento da doença.
Pessoas de baixa estatura estão em risco.
Doença de Erb-Roth e sua forma juvenil
Esta doença autossômica recessiva se desenvolve em pacientes após os 20 anos de idade, e a forma juvenil ocorre em crianças e adolescentes de 11 a 13 anos. A progressão da doença ocorre de forma ascendente, ou seja, os músculos das extremidades inferiores são afetados primeiro e a doença ascende gradativamente para as extremidades superiores. 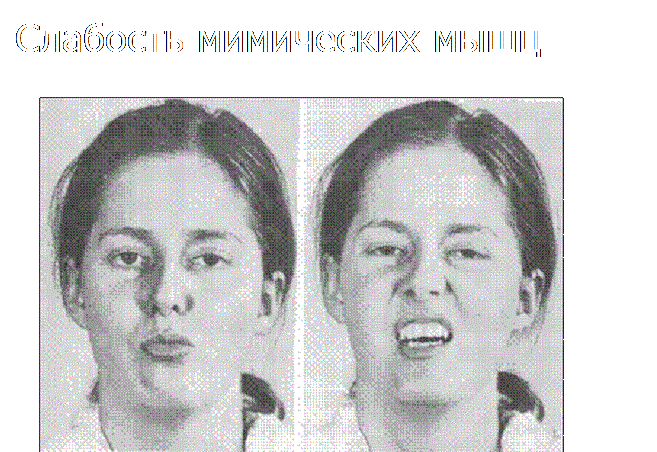
Uma característica distintiva da doença é a presença de omoplatas salientes, que se tornam mais pronunciadas e evidentes à medida que progride.
Ao caminhar, o paciente cambaleia, o abdômen se projeta e o tórax se move para dentro.
Doença de Landouzy-Dejerine
A forma escapuloumeral-facial desta doença é a mais indiscriminada, pois atinge pessoas de cinco a 55 anos. Esta doença é caracterizada por um desenvolvimento muito longo, o paciente pode permanecer capaz de trabalhar por até 25 anos de vida com esta doença.
Os sintomas distintivos são lesões nos músculos faciais, em que o paciente pode ter problemas de clareza de pronúncia devido ao fechamento incompleto dos lábios. Além disso, observa-se fechamento incompleto das pálpebras.
À medida que o paciente se desenvolve, os músculos faciais, músculos dos ombros, membros e tronco atrofiam.
Esta forma não depende de uma mutação genética.
Miopatia alcoólica
Esse tipo de doença também não está associado a mutações genéticas e há apenas um motivo para sua ocorrência - o abuso de álcool. Os pacientes podem apresentar síndromes de dor nos membros associadas especificamente a danos musculares.
Existem miopatia alcoólica aguda e crônica.
Forma distal
A distrofia muscular distal é uma variante benigna da distrofia progressiva. Via de regra, esta doença é difícil de diferenciar da amiatrofia neural de Marie-Charcot. Para separar essas duas doenças, é necessário um eletroencefalograma da cabeça, que dá uma ideia de qual doença está sendo tratada.
Os principais sintomas da doença incluem atrofia dos músculos dos membros com subsequente emagrecimento. É possível paresia dos pés, mãos, etc.
Distrofia muscular de Emery-Dreyfus
Este tipo de doença inicialmente não foi identificado como uma doença separada, uma vez que os seus sintomas eram semelhantes aos da distrofia de Duchenne. Porém, posteriormente, como resultado de um estudo de longo prazo, descobriu-se que a doença de Emery-Dreyfus apresenta sintomas individuais.
A doença é classificada como rara. Pessoas com menos de 30 anos estão em risco, geralmente jovens. Há evidências de manifestação de sintomas mesmo após 30 anos, mas são isolados.
A principal diferença entre esse tipo de doença são os problemas nos músculos do coração, que podem causar a morte. A cardiomiopatia com esta doença não é a única diferença. Além dos problemas cardíacos, os pacientes apresentam sinais padrão da distrofia de Duchenne, mas em um modo de desenvolvimento mais suave.
Causas
O aspecto negativo das doenças do sistema nervoso é que são difíceis de estudar. Por esse motivo, não se conhece totalmente os fatores que provocam o desenvolvimento de uma ou outra forma de distrofia muscular.
A principal razão para a formação da maioria dos subtipos desta doença é uma mutação genética, em particular o gene responsável pela síntese proteica.
Por exemplo, a doença de Duchenne está diretamente relacionada a uma mutação do cromossomo X sexual. Os principais portadores do gene ruim são as meninas que, apesar de sua presença no próprio DNA, não sofrem desta doença.
Quanto à forma miotônica, o culpado pela sua ocorrência é um gene localizado no cromossomo 19.
Principais sintomas
A presença de um grande número de diferentes subtipos desta doença indica diferenças nos sintomas, no entanto, a doença apresenta sintomas comuns, que incluem:

Diagnóstico de distrofia muscular
As medidas de diagnóstico para tal doença são extensas, uma vez que existe um grande número de doenças das quais é necessário diferenciar a doença.
Na fase inicial, o médico examinará definitivamente o histórico médico do paciente, esclarecerá os sintomas, a rotina diária, etc. Esses dados são necessários para traçar um plano de medidas diagnósticas subsequentes, que podem incluir:

Vale ressaltar que quanto mais tarde a doença se manifestar, melhor para o paciente, pois os primeiros sintomas na maioria dos casos terminam em morte.
Tratamento
O tratamento da distrofia muscular é um processo complexo e demorado, porém, atualmente não foi criado nenhum medicamento que cure completamente o paciente. Todas as medidas visam facilitar a vida do paciente e restaurar algumas habilidades perdidas.
Para retardar o desenvolvimento da doença, são prescritos os seguintes medicamentos:
- corticosteróides;
- vitaminas B1;
- trifosfato de adenosina (ATP).
Além disso, para desacelerar o processo de desenvolvimento, são utilizadas células-tronco fetais, que retardam o processo de degeneração.
Além disso, são prescritos como medidas preventivas:
- massagem;
- fisioterapia;
- exercícios de respiração.
Além das opções de tratamento padrão, é importante orientar-se constantemente por três componentes principais do processo de vida:
- Atividade física adequada.
- Apoio psicológico oportuno.
- Dieta.
Atividade física
A falta de vontade de uma pessoa de lutar contra uma doença tem um impacto negativo no corpo. Julgue por si mesmo, a passividade e a relutância em se mover deprimem o sistema muscular já danificado. Os músculos precisam receber uma carga, pois sem carga os processos degenerativos começam a ocorrer mais rapidamente, progredindo em ritmo mais acelerado.
A atividade física moderada e o uso de dispositivos de suporte serão uma excelente ajuda no combate à doença.
Se você tem dores musculares, natação, ioga e exercícios de alongamento são perfeitos.
Apoio psicológico
O apoio psicológico do meio ambiente é importante para uma pessoa doente. E se a doença for tão grave como esta, ainda mais. Para alguns, o apoio normal de amigos e familiares será suficiente, enquanto outros podem necessitar de ajuda psicológica qualificada.
É importante deixar claro para essa pessoa que ela não fica sozinha com seu problema. Ele deve entender que tem alguém a quem recorrer, há pessoas que o têm empatia e o apoiam.
Dieta
Quando se trata de dieta e dieta alimentar, existe uma crença comum de que seguir uma dieta antiinflamatória pode retardar a progressão da doença. Esta dieta reduz a inflamação, reduz os níveis de glicose, remove toxinas do corpo e nutre-o com nutrientes.
A essência desta dieta é a seguinte:
- Recusa de alimentos que contenham gorduras “ruins” e substituição por “boas”, introduzindo na dieta gorduras insaturadas, encontradas no azeite, na linhaça, no óleo de gergelim e no abacate.
- O uso de carne e peixe em cuja produção não foram utilizados antibióticos ou hormônios.
- Remoção completa de açúcar refinado e glúten da dieta.
- Comer os seguintes alimentos – couve chinesa, brócolis, aipo, abacaxi, salmão, beterraba, pepino, gengibre, açafrão e outros alimentos que possuem propriedades antiinflamatórias.
- Os produtos lácteos só são permitidos à base de leite de ovelha e cabra.
- É aceitável beber chás de ervas, limonada, kvass, sucos de frutas e sucos naturais.
Prevenção
Como a distrofia muscular é bastante difícil de prever e detectar numa fase inicial, as medidas preventivas resumem-se a duas recomendações simples:
Exame obrigatório de uma mulher na fase de planejamento da gravidez para presença de mutações em genes
Se, por algum motivo, um exame genético não foi feito antes da gravidez, é realizado um teste durante a gravidez para determinar mutações no cromossomo X do feto.
Prognóstico e complicações
Dependendo do tipo de doença, o prognóstico pode ser diferente, mas mesmo assim podem ser identificadas várias complicações possíveis que surgem desta doença.
- distúrbios cardíacos
- raquiocampsis
- diminuição das habilidades intelectuais do paciente
- diminuição da atividade física
- distúrbios do sistema respiratório
- resultado letal
Portanto, a distrofia muscular é uma doença bastante perigosa e incurável, por isso os futuros pais precisam ser profundamente responsáveis ao planejar uma gravidez. Cuide de você e de seus futuros filhos!





